|
В.А. Карлов
Кафедра неврологии и нейрохирургии Московского государственного медико-стоматологического университета
В начало...
(Окончание)
По механизму отрицательной обратной связи через указанные структуры ЭО оказывается подавляющее влияние на эпилептогенез, в результате чего он может быть полностью блокирован.
Баланс между эпилептической системой и системой противоэпилептической защиты в значительной степени зависит от базового функционального состояния мозга - баланса активирующих и дезактивирующих (синхронизирующих) влияний. Недостаточность активирующих систем проявляется главным образом во II стадии фазы медленного сна (стадия таламо-кортикального рекрутирования, стадия сонных веретен) - при эпилепсии сна, при переходе от сна к бодрствованию (<эпилепсия пробуждения>) либо в состоянии бодрствования (<эпилепсия бодрствования>).
Каким способом система противоэпилептической защиты подавляет эпилептогенез? По нашим данным, к таким механизмам прежде всего относится медленная активность.
Известно, что в классическом электроэнцефалографическом паттерне эпилепсии следующая за спайком медленная волна рассматривается как проявление антирекрутирующего, подавляющего влияния [31]. В глубоких стадиях фазы медленного сна эпилептическая активность <захлебывается> в медленной [3, 44].
Мы изучали роль медленной активности в подавлении эпилептической на модели опухолей роландовой области, дебютирующих эпилептическими припадками [10]. Как видно из рис. 2, по мере роста опухоли усиливается очаговая неврологическая симптоматика, появляются и затем нарастают общемозговые симптомы и медленная активность, а припадки и эпилептическая активность затухают. Иными словами, эпилептогенное поражение увеличивается, а эпилептические припадки и эпилептическая активность идут на убыль. Непрерывное нарастание при этом медленной активности дает основание считать, что последняя подавляет эпилептическую активность. Та-кой же вывод следует из анализа судорожного ЭС. При благоприятной динамике ЭС, как было установлено нами (см. выше), подавление припадков происходит за счет активного механизма замещения эпилептической активности мозга медленной, при неблагоприятном - за счет пассивного механизма истощения энергетических ресурсов, что проявляется на ЭЭГ заменой эпилептической активности изоэлектрической линией.
 |
| Рис. 2. Взаимоотношения очаговой неврологической симптоматики (1) с гипертензионными явлениями (2), эпилептической гиперсинхронной активностью (3) и медленной активностью (4) при опухолях центральной области мозга. |
Медленная активность может не позволить развиваться очередному припадку, подавляя его начальную стадию - ритм вовлечения.
Малоизученными в эпилептогенезе являются механизмы межполушарного уровня. Проведенное в нашей клинике специальное изучение этого вопроса позволило установить, что при правополушарном очаге генерализации эпилептической активности часто непосредственно предшествует вспышка левополушарной активности. Исследование синхронности биопотенциалов головного мозга у больных эпилепсией выявило ее нарастание по мере заболевания, преимущественно в левом полушарии [20]. Это позволило сформулировать концепцию, согласно которой при правополушарных очагах (у правшей) генерализация эпилептической активности происходит прежде всего с вовлечением генерализующихся субкортикальных структур левого полушария, тогда как при левополушарных - <своего>, а затем другого. Тем самым подтверждается выдвинутое нами ранее предположение о большей <эпилептичности> левополушарных ЭО по сравнению с правополушарными [7].
Нами сформулировано положение о том, что конечным этапом эпилептогенеза при прогредиентном течении эпилепсии является тотальная эпилептизация нейронов - <эпилептический мозг>. Под влиянием постепенного воздействия ЭО, <запускающего> по сути kindling-механизм, меняются функциональные свойства рецепторов трансмиттерных систем, синапсов и самих нейронов за пределами очага с постепенным вовлечением всего мозга. Происходит прогрессирующее усложнение эпилептической системы, вовлечение в эпилептогенез новых структур. Постепенно изменяются информационные функции нейронов вне ЭО. Последний фактически не только дезорганизует функцию мозга, но и управляет им, реорганизуя его специфическим эпилептическим образом [2, 11]. В механизме эпилептизации может иметь значение ряд механизмов, в том числе функциональное истощение интернейронов, обеспечивающих в норме селекцию поступающих импульсов [6]. Возникает постоянный самоподдерживающий процесс, связанный со значительными перестройками на биологическом и молекулярном уровнях. В дальнейшем возникает метаболическая недостаточность нейронов вследствие превышения спроса над предложением, увеличения энергетических затрат и относительной недостаточности кровообращения. Постепенно метаболическая недостаточность переходит в структурную с повреждением главным образом аксодендритических связей и преобладанием аксосоматических, характеризующихся легкостью запуска мембранных потенциалов [6]. В этом процессе принимает участие и глия, которая на первых этапах компенсирует метаболическую недостаточность нейронов путем форсирования синтеза белка для нейронов (гипертрофия глии - морфологическое проявление этого процесса); на последующих этапах происходит истощение глии и развивается прогрессирующий глиоз, что приводит к уплотнению мозговой ткани, недостаточности капиллярного кровотока и другим нарушениям [1]. При этом нарушается и аккумуляторная функция глии, в частности, при разрядке нейрона не аккумулируется калий, который, находясь в межклеточной жидкости, вызывает деполяризацию других нейронов [30].
Приведенная схема эпилептизации мозга хорошо вписывается в характеристики механизма симптоматической эпилепсии. Что касается идиопатической эпилепсии, то здесь, как известно, до последнего времени не удавалось идентифицировать ни эпилептогенного поражения, ни ЭО. Считается, что пейсмекером эпилептической активности являются неспецифические ядра зрительного бугра. В частности, в основе абсансных припадков лежит таламокортикальная петля: глутаматергические (неспецифические) пирамидные нейроны, задействованные петлей обратной связи с ГАМК-ергическими нейронами ретикулярных ядер зрительного бугра [26]. В последнее время <нащупываются> микроструктурные нарушения, которые могут лежать в основе эпилептогенеза. Чаще всего это эктопия нейронов мозговой коры, занимающих необычное положение [40, 41]. Последнее сопровождается нарушением нейрональных связей, дефицитом афферентации, развитием деафферентационной сверхчувствительности. Даже если утверждать, что пейсмекером эпилептического припадка являются неспецифические ядра зрительного бугра, неясно, как он включается при данной ситуации. Нами накоплены данные о том, что по крайней мере в части случаев при идиопатической генерализованной эпилепсии эпилептическая активность возникает в медиобазальных отделах больших полушарий головного мозга. Вначале такое утверждение основывалось на случаях, когда при классической картине типичного абсанса удавалось обнаружить очаговую эпилептическую активность на ЭЭГ и/или асимметрию латентных периодов и амплитуд компонентов зрительных вызванных потенциалов [9]. Сейчас это убеждение подкреплено материалами, полученными нами совместно с В.В. Гнездицким с помощью метода многошаговой дипольной локализации. Показана вовлеченность обширных участков мозга в генез спайк-волнового комплекса преимущественно корковых, но и с участием глубинных структур. Сделан вывод, что генерализованная билатерально-синхронная симметричная активность также может иметь фокальное происхождение - при расположении ЭО в медиальных отделах лобно-височной области или структурах таламуса с моментальной генерализацией [17]. Эта сторона проблемы требует дальнейшего изучения. В случае ее подтверждения приведенная выше схема этапов эпилептогенеза становится универсальной.
В заключение следует сказать о том, что с биологической и нейрофизиологической точек зрения есть припадок. В 1991 г. нами была представлена [12], а затем подтверждена дальнейшими публикациями [13] концепция пароксизмальных состояний в неврологии. Согласно этой концепции, любой припадок, т.е. приступ церебрального происхождения, является срывом компенсаторного напряжения гомеостатических механизмов разного уровня - в зависимости от основной патологии синаптического, нейронального, системного, межсистемного, органного и др. Этот срыв возникает под влиянием тех или иных экзогенных либо эндогенных воздействий (рисунок 3). Последствия такого срыва неоднозначны. Оставляя в стороне вопрос о патогенном влиянии припадка на мозг, отметим возможность определенных саногенных влияний.
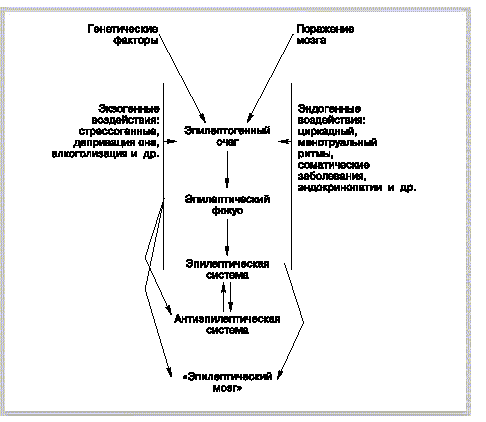 |
Рисунок 3. Развитие пароксизмальных состояний при заболеваниях нервной системы. |
На рис. 4 приведены результаты психологического обследования больного эпилепсией с частотой судорожных припадков 1-2 в месяц, у которого по мере приближения припадка ухудшалось состояние, появлялись депрессия, беспокойство, нарушалась трудоспособность. После припадка состояние восстанавливалось, нормализовались настроение и трудоспособность. Исследования проводились до и после припадка. Тестировались воспоминания из жизни больного. В
целом после приступа объем воспоминаний снижался, однако важно другое - резко менялось соотношение позитивно и негативно окрашенных воспоминаний: до припадка преобладали негативно, после припадка - позитивно окрашенные воспоминания.
 |
Рис.4. Автобиографический тест оценки депрессии. |
Это подтверждает выдвинутую нами концепцию, согласно которой припадок мобилизует компенсаторные резервы, позволяющие организму выйти из припадка и в течение того или иного времени сохранять более высокий уровень функционирования, чем до развития приступа. Вызывают удивление публикации [4], авторы которых значительно позже пришли к подобному выводу, знают о наших исследованиях, но не ссылаются на них.
Журнал неврологии и психиатрии N 9-2000, стр.7-15
1. Ахмеров Н.У. Журн невропатол и психиатр 1982; 82: 6: 106-108.
2. Бехтерева Н.П., Камбарова Д.К., Поздеев В.К. Устойчивое патологическое состояние при болезнях мозга. М: Медицина 1978.
3. Биниауришвили Р.Г., Вейн А.М., Гафуров Б.Г., Рахимджанов А.Р.
Эпилепсия и функциональные состояния мозга. М: Медицина 1985.
4. Вейн А.М., Воробьева О.В. Журн неврол и психиатр 1999; 99: 12: 8-12.
5. Власов П.Н. Восточно-Европейская конференция <Эпилепсия и клиническая нейрофизиология>: Труды. Гурзуф 1999; 18-19.
6. Зенков Л.Р. Болезни нервной системы. Под ред. Н.Н. Яхно и др. М: Медицина 1995; 1: 14-18.
7. Карлов В.А. Эпилептический статус. М: Медицина 1974.
8. Карлов В.А., Петренко С.Е. Нейрофизиологические механизмы эпилепсии. Тбилиси 1980; 221-228.
9. Карлов В.А., Овнатанов В.С. Журн невропатол и психиатр 1987; 87: 6: 805-811.
10. Карлов В.А., Бова В.Е., Маджидов Н.М. Джексоновский припа-док. Ташкент: Медицина 1988.
11. Карлов В.А. Эпилепсия. М: Медицина 1990.
12. Карлов В.А. Пароксизмальные состояния в неврологии: Материалы пленума Всесоюзного общества невропатологов и Научного совета по неврологии АМН СССР (19-21.06.91). Киев 1991; 50. 13. Карлов В.А. Журн неврол и психиатр 1993; 93: 2: 82-83. 14. Карлов В.А. Журн неврол и психиатр 1996; 96: 2: 5-8. 15. Карлов В.А., Хабибова А.О. VI Российский национальный конгресс <Человек и лекарство>: Тезисы докладов. М 1998; 95. 16. Карлов В.А. Журн неврол и психиатр 1999; 99: 5: 3-8.
17. Носкова Т.Ю., Гнездицкий В.В., Карлов В.А., Пирник Г.А. Восточно-Европейская конференция <Эпилепсия и клиническая ней-рофизиология>: Труды. Гурзуф 1999; 30-32.
18. Пенфилд У., Джаспер Г. Эпилепсия и функциональная анатомия головного мозга человека. М 1958.
19. Сараджишвили П.М., Геладзе Т.Ш. Эпилепсия. М: Медицина 1977. 20. Селицкий Г.В., Карлов В.А., Сорокина Н.Д. Журн высш нерв деят 1995; 1: 78-89.
21. Степанова Т.С., Грачев К.В., Шустин В.А. Хирургическое лечение эпилепсии. Тбилиси 1985; 139-141.
22. Терминологический словарь по эпилепсии. ВОЗ. Женева 1975; 32. 23. Терминологический словарь по эпилепсии. ВОЗ. Женева 1975; 73. 24. Терминологический словарь по эпилепсии. ВОЗ. Женева 1975; 24-25.
25. Шандра А.А., Годлевский Л.С., Брусенцов А.И. Киндлинг и эпилептическая активность. Одесса: Астра Принт 1999.
26. Clark S., Wilson W. The Treatment of Epilepsy: Prnciples end Practice. Second edition. Ed. E. Wyllie 1997; 53-81.
27. Commission on Classification and Terminology of the International League Against Epilepsy. Epilepsia 1981; 22: 489-540.
28. Commission on Classification and Terminology of the International League Against Epilepsy. Epilepsia 1989; 30: 189-199.
29. Engel J., Kuhl D.E., Phelps M.E. Science 1982; 218: 64-66. 30. Fisher R.S. Epilepsy, 2nd ed. Ed. A. Hopkins et al. London and all 1995; 35-58.
31. Gastaut H., Fischer-Willams M. Handbook of Neurophysiology. Washington 1959; 1: 329-363.
32. Gowers W.R. Epilepsy and other Chronice Convulsive Diaseаses.
London 1881.
33. DeLorenz R.J., Jarhette L.-K., Towne A.R. Epilepsia 1999; 40: 164- 169.
34. Hagemann G., Bruehl C., Lutrennberg M. et al. Epilepsia 1998; 39: 339-348.
35. Johnson M.V. Treatment of Epilepsy: the Principles and Practics: 2nd ed. Ed. Wyllie. Baltimor and all 1997; 122-150.
36. Karlov V.A. Epilepsia 1998; 39: 2: 32.
37. Keranen P., Sillanpaa M., Riekinnepy J. Epilepsia 1988; 29: 1-7. 38. Lennox W.J. Epilepsy and Related Disorders. Boston, Mass. Little Brown and Co 1960.
39. Lowenstein D.H., Bleck T., Mac Donald R.L. Epilepsia 1999; 40: 120- 122.
40. Meencke H.J., Janz D. Epilepsia 1984; 25: 8-21. 41. Meencke H.J. Epilepsia 1999; 40: 2: 74.
42. Morrel M. The Treatment of Epilepsy: the Principles and Practice: 2nd ed. Ed. Wyllie. Baltimor and all 1997; 179-190.
43. Patsalos Ph.N. Women end Epilepsy. Ed. M.R. Trimble. Chichester and all. 1991; 135-149.
44. Rossi G.F., Collicchio G., Pola P., Rosseli R. Sleep Deprivation. Ed.
R.Degen, E.A. Rodin. Amsterdam and all 1991; 23.
45. Sander J.W., Hart Y.M., Jonson A.L., Shorvon S.D. Lancet 1990; 336: 1267-1976.
46. Shorvon S. Epilepsy: 2nd ed. Ed. A. Hopkins et al. London a all 1995; 331-354.
47. Spenser S.S. Epilepsia 1994; 34: 6: 72-89. 48. Spenser S.S. Epilepsia 1998; 38: 114-119.
Написать комментарий
| 