|
Е.Д. Белоусова, М.Ю. Никанорова, Е.А. Николаева Московский НИИ педиатрии и детской хирургии Минздрава РФ
В начало...
Статья посвящена актуальной и малоизвестной широкому кругу врачей проблеме - диагностике и лечению врожденных дефектов метаболизма в неонатальном периоде. Описываются симптомы, на основании которых педиатр и невролог могут заподозрить у новорожденного метаболическое заболевание, приводится алгоритм необходимых лабораторных исследований. Кратко изложены общие принципы лечения неонатальных врожденных дефектов метаболизма.
Ключевые слова:
Диагностика и лечение врожденных аномалий метаболизма - одна из самых сложных задач как для невролога, так и для педиатра. С одной стороны, трудности диагностики связаны с клиническим полиморфизмом заболеваний, по-разному протекающих у детей различного возраста, с другой - с тем, что самые различные нарушения обмена в одной и той же возрастной группе могут иметь сходные клинические проявления [1].
Особые трудности вызывает диагностика врожденных дефектов метаболизма у новорожденного [2]. Заболевания протекают тяжело и заканчиваются летально до того, как становится очевидной специфическая метаболическая и неврологическая картина. В периоде новорожденности проявляются примерно 25% известных наследственных болезней обмена веществ и почти все они отличаются особой тяжестью состояния ребенка. По остроте начала и течению наследственные болезни обмена веществ в неонатальном периоде могут напоминать нейроинфекции и нейродистресс-синдром. Однако явная прогредиентность течения с утратой функций не характерна для новорожденного, так как сами эти функции еще не сформированы. Поэтому большинство детей с неонатальными формами наследственных дефектов метаболизма либо умирают в первые месяцы жизни без установленного диагноза, либо длительно наблюдаются с самыми разными неадекватными диагнозами. Чаще всего наследственные болезни обмена скрываются под маской детского церебрального паралича, умственной отсталости и эпилепсии, резистентной к антиконвульсантам. Причины неонатальных наследственных метаболических нарушений многочисленны (см. ниже).
Нарушения метаболизма аминокислот
Нарушения цикла синтеза мочевины (дефицит N-ацетилглутаматсинтетазы, карбамоилфосфатсинтетазы, орнитинкарбамоилтрансферазы, аргининсукцинатсинтетазы, аргининсукцинатлиазы и аргиназы) Гиперорнитинемия - гипераммониемия - гомоцитруллинурия
Болезнь с запахом мочи кленового сиропа (лейциноз)
Нарушения метаболизма органических кислот
Некетотическая гиперглицинемия
Изовалериановая ацидемия
Пропионовая ацидемия
Метилмалоновая ацидемия
Мевалоновая ацидурия
3-гидрокси-3-метилглутаровая ацидемия
Митохондриальные энцефалопатии
Нарушения в системе пируватдегидрогеназного комплекса (дефицит пируваткарбоксилазы, пируватдегидрогеназы, дигидролипоилтрансацетилазы и дигидролипоилдегидрогеназы) Нарушения функций дыхательной цепи (дефициты ферментов I-IV комплекса дыхательной цепи) Нарушения функций ферментов цикла Кребса (дефицит фумаразы, сукцинатдегидрогеназы,  -кетоглутаратдегидрогеназы и аконитазы) Нарушения синтеза и обмена карнитина (системная недостаточность карнитина, дефицит карнитинпальмитоилтрансферазы II и ацилкарнитинтранслоказы) Нарушения -кетоглутаратдегидрогеназы и аконитазы) Нарушения синтеза и обмена карнитина (системная недостаточность карнитина, дефицит карнитинпальмитоилтрансферазы II и ацилкарнитинтранслоказы) Нарушения 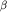 -окисления жирных кислот (дефициты ацил-СоА-дегидрогеназ жирных кислот с короткой, средней, длинной и очень длинной углеродной цепью, множественный дефицит ацил-СоА-дегидрогеназ) -окисления жирных кислот (дефициты ацил-СоА-дегидрогеназ жирных кислот с короткой, средней, длинной и очень длинной углеродной цепью, множественный дефицит ацил-СоА-дегидрогеназ)
Нарушения обмена биотина
Дефицит биотинидазы
Дефицит синтетазы голокарбоксилаз
Болезни пероксисом
Синдром Цельвегера
Неонатальная адренолейкодистрофия
Гиперпипеколовая ацидемия
Дефицит ацил-СоА-оксидазы пероксисом
Дефицит 3-оксиацил-СоА-тиолазы пероксисом
Бифункциональная недостаточность протеинов
Лейкодистрофии
Болезнь Краббе
Лейкодистрофия Канавана - Ван Богарта - Бертранда
Метахроматическая лейкодистрофия
Лейкодистрофия Пелицеуса - Мерцбахера
Суданофильные лейкодистрофии
Нарушения обмена углеводов
Дефицит фруктозо-1,6-альдолазы
Наследствення непереносимость фруктозы
Галактоземия (дефицит галактозо-1-фосфатуридилтрансферазы)
Редко, но встречаются неонатальные формы таких заболеваний, как болезнь Менкеса, гликогеноз типа II, болезнь Нимана-Пика и Гоше, синдром Барта, тирозинемия I типа, болезнь Альперса, а также сиалидоз [3].
Эпидемиология. Данные о распространенности наиболее изученных наследственных метаболических заболеваний приведены в табл. 1.
| Таблица 1. Распространенность наследственных заболеваний обмена веществ с неонатальным дебютом |
| Заболевание |
Частота |
| Дефицит аргининсукцинатлиазы |
1/70 000 |
| Дефицит карбамоилфосфатсинтетазы |
1/1 000 000-1/800 000 |
| Дефицит аргининсукцинатсинтетазы |
1/250 000 |
| Некетотическая гиперглицинемия |
1/55 000 |
| Болезнь с запахом мочи кленового сиропа |
1/200 000 |
| Изовалериановая ацидемия |
1/200 000 |
| Пропионовая ацидемия |
1/350 000 |
| Метилмалоновая ацидемия |
1/48 000 |
| Дефицит ацил-СоА-дегидрогеназы жирных кислот со средней длиной углеродной цепи |
1/9 000-1/13 000 |
| Синдром Цельвегера |
1/50 000-1/100 000 |
| Дефицит биотинидазы |
1/35 000-1/54 000 |
| Дефицит орнитинкарбамоилтрансферазы |
1/80 000 |
| Дефицит аргининсукцинатлиазы |
1/90 000 |
| Тирозинемия I типа |
1/30 000 |
| Аргининемия |
1/990 000 |
Распространенность других, менее изученных дефектов метаболизма неизвестна. Число описаний больных с редкими неонатальными метаболическими заболеваниями колеблется от единичных до сотен наблюдений.
Генетические данные. Большинство врожденных дефектов метаболизма с проявлениями в неонатальном периоде имеют аутосомно-рецессивный тип наследования. Исключениями из правила являются неонатальная адренолейкодистрофия, лейкодистрофия Пелицеуса-Мерцбахера, болезнь Менкеса и синдром Барта, которые наследуются по рецессивному сцепленному с Х-хромосомой типу, и дефицит орнитинкарбамоилтрансферазы, который передается доминантно, в сцеплении с Х-хромосомой. Дефицит пируватдегидрогеназы и множественный дефицит ацил-СоА-дегидрогеназ имеют два возможных типа наследования - аутосомно-рецессивный и рецессивный, сцепленный с Х-хромосомой. Нарушения в системе транспортной цепи электронов в дыхательной цепи наследуются по митохондриальному типу (материнское наследование). Дефицит I комплекса дыхательной цепи (NADH; CoQ-редуктазы) наследуется как по аутосомно-рецессивному, так и по митохондриальному типу, описаны также X-cцепленные формы.
Значительные успехи достигнуты в молекулярной диагностике неонатальных дефектов метаболизма. В табл. 2 представлены врожденные метаболические заболевания с картированными генами.
| Таблица 2. Локализация генов, ответственных за развитие наследственных заболеваний обмена веществ с неонатальным дебютом |
| Заболевание |
Хромосома, локус |
| Болезнь с запахом мочи кленового сиропа: |
| -ген Е1 |
19q13.1-q13.2 |
| -ген Е1 |
6p22-p21 |
| -ген Е2 |
1p31 |
| -ген Е3 |
7 |
| Изовалериановая ацидемия |
15q13-q15 |
| Пропионовая ацидемия: |
| --cубъединица |
13q32 |
| --субъединица |
3q21-q22 |
| Метилмалоновая ацидемия |
6р21.2-р12 |
| Дефицит пируваткарбоксилазы |
11 (длинное плечо) |
| Дефицит пируватдегидрогеназы: |
| --cубъединица |
Хр22-сеп |
| --субъединица |
3 |
| Дефицит дегидролипоилдегидрогеназы |
7q31-q32 |
| Дефицит IV комплекса дыхательной цепи: |
| -4-я субъединица |
16q22 |
| -Vb-cубъединица |
2сеп-q13 |
| Дефицит фумаразы |
1q42.1 |
| Дефицит сукцинатдегидрогеназы |
1р35-р36.1 |
| Дефицит карнитинпальмитоилтрансферазы II |
1р11-р13 |
| Синдром Барта |
Хq28 |
| Болезнь Менкеса |
Хq13.3 |
| Некетотическая гиперглицинемия |
9 (короткое плечо) |
| Дефицит ацил-СоА-дегидрогеназы жирных кислот: |
| -со средней длиной углеродной цепи |
1р22.1 |
| -с короткой углеродной цепью |
12 |
| -с длинной углеродной цепью |
2q34-q35 |
| Дефицит биотинидазы |
3р25 |
| Дефицит синтетазы голокарбоксилаз |
21q22.1 |
| Синдром Цельвегера |
8q21 |
| Болезнь Kраббе |
14q21-q31 |
| Лейкодистрофия Kанавана - Ван Богарта - Бертранда |
17р13-pter |
| Метахроматическая лейкодистрофия |
22q13.31-qter |
| Лейкодистрофия Пелицеуса-Мерцбахера |
Хq22 |
| Множественный дефицит ацил-СоА-дегидрогеназ (дефицит электроннотранспортного флавопротеина) |
15q23-25 |
Далее...
Написать комментарий
| 