ГЛАВА 2
ВАЖНЕЙШИЕ РАЗРАБОТКИ В ТЕОРИИ ГИПЕРГЕНЕЗА ВО ВТОРОЙ ПОЛОВИНЕ XX ВЕКА
В последние десятилетия ХХ в. генетическая концепция гипергенеза пополнилась рядом новых разработок, которые оказались сосредоточенными на ее начальном этапе. Начало всегда является особенно важным, как непосредственно определяющее течение событий не только в ближайшем, но и в дальнейшем будущем. В данном случае оно соотносится с механизмами деструкции исходных (первичных) минералов и в соответствии с главенствующей в ХХ в. гидрохимической моделью гипергенного процесса - с взаимодействием природных водных растворов с первичными минералами, рудами и породами.
К определенным достижениям в этом плане, видимо, следует отнести результаты следующих исследований:
I. Становление коррозионной модели развития гипергенеза;
II. Вывод корректных уравнений реакций окисления минералов;
III. Выявление количественных причинно-следственных связей в "цепочке" факторов: геоморфология региона - динамика подземного стока - химизм и pH вод - зональность и тип гипергенной минерализации;
IV. Закон энергетической направленности гипергенных процессов;
V. Обоснование роли бактериального фактора в развитии гипергенеза.
Остановимся подробнее на первых трех пунктах приведенного перечня.
I. Становление коррозионной модели в теории гипергенеза произошло в 1970-е годы благодаря многочисленным экспериментальным работам и обобщениям, проводимым на кафедре минералогии МГУ (Л. К. Яхонтова, А. П. Грудев, Л. Г. Нестерович). Эти исследования осуществлялись в рамках классической гидрохимической модели, опирающейся на значение роли в гипергенезе взаимодействия природных растворов с подвергающимися выветриванию минералами, в условиях критического отношения к распространенным в те годы представлениям о первостепенном значении в образовании гипергенных минералов, якобы накапливающихся и протекающих в земной коре электрических токов (Яхонтова, Грудев, 1978, 1987).
Коррозионная модель процессов гипергенеза сложилась в условиях электрохимических исследований арсенидов, сульфоарсенидов и многих сульфидов. Эти исследования показали, что деструкция минералов осуществляется по законам коррозионных или анодно-катодных процессов, когда под воздействием окисляющей среды (электролита) погруженный в нее минерал-электрод, попадая в анодную позицию, подвергается анодному окислению, а катодный процесс развивается в среде, например на растворенном в ней кислороде: O2+2H2O+4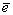 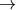 4(OH) или на другом минерале, играющем роль катода.
4(OH) или на другом минерале, играющем роль катода.
Катодно-анодная функция минерала связана с его кристалло-структурными особенностями, описываемыми, как известно, теорией зонного строения прежде всего полупроводников - малая ширина запрещенной зоны, облегчая выход электрона в зону проводимости кристалла, т.е. уменьшая "работу выхода", является залогом его анодной позиции в катодно-анодном взаимодействии. Минерал, погруженный в электролит, характеризуется специфическим электродным потенциалом (ЭП) и в таком взаимодействии занимает ту или иную позицию: в анодной окисляется с потерей электронов и является их донором, в катодной - становится устойчивым акцептором. Отсюда катодно-анодные отношения минералов в зоне окисления могут быть оценены как донорно-акцепторные.
В горизонтах цементации гипергенных зон рудных месторождений, где господствуют застойные воды, обедненные кислородом и другими окислителями, электрохимические донорно-акцепторные взаимодействия между минералами и средой практически сводятся к нулю. Об этом свидетельствуют опыты с вытеснением в электролите воздуха (гелием), когда среда теряла свой окислительный потенциал (ОП) и сравнивалась с ЭП использованного в опытах минерала В этих случаях на первое место выходят обменные реакции образования цементационных сульфидов и биоминеральные (биокосные) процессы с участием сульфатредуцирующих микроорганизмов (Яхонтова, Грудев, 1974).
Следует заметить, что биокосные взаимодействия полностью вписываются в рамки коррозионной модели развития начального этапа гипергенеза. Микроорганизмы в горизонтах просачивания вод служат, как правило, "катодным" партнером минералов, т.е. агрессивным окислителем, акцептирующим электроны окислительных реакций. Становление коррозионной модели развития зоны гипергенеза способствовало описанию протекающих в ней окислительно-восстановительных реакций не традиционными с балансом вещества и зарядов (валентностей) уравнениями, а ионно-электронными реакциями.
Новая концепция формирования гипергенной зоны стала методологической основой для углубленного исследования механизмов окисления минералов. Появилась реальная экспериментальная возможность проникнуть в сущность процессов, протекающих непосредственно в область электролита (раствора), примыкающего к исходной поверхности окисляющегося минерала, - в так называемый двойной электрический слой (ДЭС), где происходит концентрация первых анионных продуктов окисления и где совершается ряд специфических первых же реакций (диффузия, сорбция и десорбция, массообмен и др.), во многом определяющих течение собственно деструкционных механизмов.
Коррозионная модель формирования гипергенной зоны особо плодотворной оказалась при исследовании минералов, контактирующих друг с другом, - гальванических пар. Она позволила, по-существу, количественно оценить химизм деструктирующих процессов, кинетику и энергетику взаимодействий, специфику протекания окислительных реакций в условиях массивных и вкрапленных руд, рассмотреть проблему устойчивости минералов, наконец, определиться в отношении роли бактериального фактора в гипергенезе.
Знаменательно, что новая генетическая разработка существенно увеличила приток в генетические проблемы гипергенной минералогии современных достижений физической химии, учения о растворах, энергетической кристаллохимии, электрохимии и даже микробиологии. Не отрекаясь от классического канона о теснейшей связи результатов гипергенеза с гидрохимическими, тектоническими и климатическими факторами и, наоборот, опираясь на них, она внесла новые мировоззренческие представления в генетические проблемы гипергенной минералогии.
II. В последних десятилетиях века общепризнанной необходимостью стал поиск способов составления уравнений, отражающих наиболее вероятные реакции окисления минералов. Коррозионная модель формирования гипергенной зоны позволила подойти к решению такой задачи с опорой на количественные экспериментальные данные и современное состояние важнейших разделов физической химии неорганических систем-равновесий в водных растворах и кинетики протекающих реакций.
Как отмечалось, окисление отдельных минералов согласно гидрохимической модели развития гипергенеза долгое время описывалось реакциями, в которых господствовало главное правило химической стехиометрии левой и правой частей уравнений окисления типа MeS+2O2=FeSO4 или FeS2+3,5O2+H2O=FeSO4+H2SO4 (Смирнов, 1951). Лишь в результате исследований Р. Гаррелса (1960-е годы) в обиход вошли диаграммы Eh-pH и ставшие хорошо известными ионно-электронные уравнения, описывающие и коррозию металлов и окисление сульфидов. Так, окисление того же пирита могло быть записанным новой реакцией FeS2+3,25O2+1,5H2O=Fe3++2HSO4-+H++2, но вновь с произвольной трактовкой правой части. Возникла необходимость обоснованной записи уравнений окислительно-восстановительных реакций, отвечающей более реальной картине в отношении состояния вещества (Fe, S) в определяющих это состояние координатных Eh-pH условиях.
Эту задачу удалось решить с помощью несложного эксперимента, в первую очередь способного определить зависимость ЭП-pH сульфидного электрода в сернокислом электролите с постепенным увеличением pH и довести полученный график до представительного путем двух-трех повторов опыта. В координатах Еh-pH в таком графике могут быть три главных позиции отдельных его участков (рис.1): 1) участок А, отвечающий реакциям типа гидролизных, независимых от изменения ЭП и содержащих в продуктах реакции не электроны , а ионы H+; 2) Участок В, связанный с содержанием в продуктах реакции лишь электронов; 3) Участок С - наклонный, отражающий окислительно-восстановительный процесс, на котором правая часть уравнений содержит и H+ и . Это наиболее сложный вариант выбора типа реакции.
Согласно известному уравнению Нернста, отражающему зависимость ЭП от активности ионов, участвующих в потенциалопределяющих реакциях, 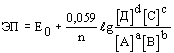 (для реакции аА+bB
dД+сС+n, в которой Д или С могут быть ионы H+), можно убедиться в том, что важнейшее частное от деления (для реакции аА+bB
dД+сС+n, в которой Д или С могут быть ионы H+), можно убедиться в том, что важнейшее частное от деления 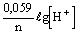 определяет отношение количества ионов H+ (pH) к числу (Eh) в продуктах окислительной реакции, позволяющее в правой части уравнения записывать не любое число H+ и , а только диктуемое экспериментом - наклоном графика ЭП-pH в рассматриваемом участке изменения pH. Наклон графика в виде tg угла наклона измеряется экспериментально и с учетом множителя 0,059(0,06) может быть: [-0,010] - H+ и 6; [-0,015] - H+ и 4; [-0,060] - H+ и (для того же пирита при pH<3); [-0,020] - 3H+ и 4 (для Bi2S3) и т.д. Благодаря описанным приемам количество ионов H+ и в продуктах реакции не может быть записано произвольно, а лишь с сохранением между ними экспериментального отношения. определяет отношение количества ионов H+ (pH) к числу (Eh) в продуктах окислительной реакции, позволяющее в правой части уравнения записывать не любое число H+ и , а только диктуемое экспериментом - наклоном графика ЭП-pH в рассматриваемом участке изменения pH. Наклон графика в виде tg угла наклона измеряется экспериментально и с учетом множителя 0,059(0,06) может быть: [-0,010] - H+ и 6; [-0,015] - H+ и 4; [-0,060] - H+ и (для того же пирита при pH<3); [-0,020] - 3H+ и 4 (для Bi2S3) и т.д. Благодаря описанным приемам количество ионов H+ и в продуктах реакции не может быть записано произвольно, а лишь с сохранением между ними экспериментального отношения.
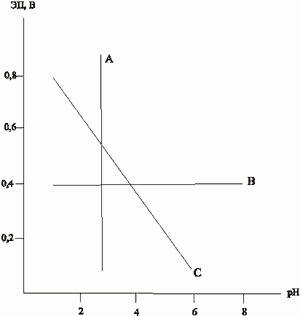
Рис. 1.Возможное положение отдельных участков графиков ЭП-pH минерального электрода в окисляющем растворе. Пояснение см. в тексте.
Рассмотрим некоторые примеры:
1. Реакция Fe2+
Fe3++ - наклон [O];
2. Окисление халькопирита - на графике ЭП-pH выявлено два участка: 1) в области pH<3 c наклоном [-0,020] и 2) в области с pH 3-8 c наклоном [O]. Выводятся два уравнения окисления сульфида:
[-0,020]CuFeS2+3,25O2+1,5H2O
[CuHSO4]++[FeHSO4]++H++3 и
[O] CuFeS2+4H2O
[CuSO4]0+[FeSO4]++.
Один из наиболее сложных вопросов в практике вывода уравнений окисления минералов связан с поиском данных о состоянии вещества в растворах в тех или иных условиях состояния среды. Для корректных результатов недостаточно пользоваться известными диаграммами Eh-pH, предлагаемыми М. Пурбэ (Pourbaix, 1963), Р. Гаррелсом и Ч. Крайстoм (1968), - необходим тщательный анализ литературы и экспериментально подтвержденных сведений. Проблема осложняется, когда окислению подвергаются контактирующие друг с другом минералы или находящиеся в контакте с микроорганизмами.
Корректные уравнения окисления минералов становятся прогнозом реального состояния продуцируемых процессом их деструкции растворов, миграции последних и характера их участия в гипергенной и техногенной минерализации, а также в экологических последствиях.
Описанный метод вывода уравнений окисления минералов полностью применим и к случаям исследования процессов окисления минералов в культуральных (бактериальных) средах, которые являются более агрессивными, ведущими процесс с максимально высоким выходом энергии (электронов) в среду, обеспечивающей жизнедеятельность микроорганизмов. Понятно, что в этих случаях графики ЭП-pH существенно изменяют свой вид, и в соответствии с чем, и выведенные для них уравнения реакций окисления минералов. Полученные при этом данные имеют прямое отношение к организации биотехнологической переработки минерального сырья, а разработанная и описанная методика служит основой ее совершенствования.
Третья новейшая разработка, которую также хотелось отметить, являясь существенным вкладом в теорию гипергенеза, раскрывает прямую связь химизма и щелочности-кислотности природных вод с динамикой их просачивания и истечения во вмещающих породах и в конечном счете - с геоморфологическими особенностями региона. В силу обстоятельств эти новейшие исследования оказались в большей мере соотносящимися с развитием кор выветривания, но их значение остается не менее важным и для формирования минерализации окисленных руд.
Таблица 31
Связь состояния растворов и корообразования с геоморфологией
региона по данным Ю. Ю. Бугельского (1979)
|
Характер растворов |
Вершина (очень сильный водообмен) |
Пологий склон (средний водообмен) |
Плато (слабый водообмен) |
|
К* |
|
30 |
15 |
0,5 |
|
Mg |
мг/л |
7-8 |
40-80 |
80-150 |
|
SiO2 |
мг/л |
10-15 |
100-130 |
80-100 |
|
HCO3 |
мг/л |
до 100 |
до 300 |
до 600 |
|
pH |
|
6,5 |
7-8 |
8-9 |
|
Рельеф
|

|
|
Тип профиля выветривания |
Резко сокращенный профиль (дезинтеграция) |
Полный профиль (с горизонтом нонтронитизации) |
Недоразвитый профилъ (с желез-ной шляпой) |
*Коэффициент подземного стока, л/сек/км2.
В табл. 31, показано, как тесно увязаны водообмен (коэффициент подземного стока К), химический состав (содержание в водах Mg, SiO2 и HCO3-) и величина pH просачивающихся через толщу пород (в данном случае змеевиков) растворов с рельефом местности и сколь различны результирующие типы и профили коры выветривания, отвечающие привершинной части, пологим склонам и плато рельефа местности.
Следует отметить, что эта ювелирная работа выполнена на Кубе
Ю. Ю. Бульским - руководителем экзогенного отдела ИГЕМа АНРФ, сотрудники которого
(В. М. Новиков, А. Д. Слукин, Н. Б. Сергеев) продолжают детальнейшие исследования
минерализации кор выветривания и ее связей с динамикой и химизмом природных
вод, климатической и тектонической обстановкой. Отрадно отметить, что в последние
годы подобные тонкие исследования были распространены и на окисленные руды
(Сергеев, 1995; Сергеев и др., 1996).
|