|
Е.А. Николаева, Л.З. Казанцева, П.А. Темин, М.Ю. Никанорова, М.Б. Курбатов, В.С. Сухоруков, М.В. Прыткина
Представлено описание ребенка и его матери, страдающих митохондриальным синдромом MERRF. Проявления болезни у ребенка характеризовались тяжестью, упорным прогрессированием, положительной динамикой в процессе лечения. Клиническая картина заболевания у матери отличалась сочетанием типичной симптоматики с необычными проявлениями в виде бородавчатого невуса и обширной гемангиомы.
Ключевые слова:
Заболевания, сопровождающиеся тяжелым поражением нервной системы, занимают особое место в патологии детского возраста, в значительной степени определяя медицинскую и социальную дезадаптацию детей. Среди причин, детерминирующих генез этих состояний, в последние годы выделяют группу митохондриальных болезней. Тяжелое прогрессирующее течение большинства этих состояний обусловливает высокую степень инвалидности больных и нередко приводит к их гибели [1]. Основу патогенеза митохондриальных заболеваний составляет нарушение внутримитохондриальных процессов высвобождения и утилизации энергии, кодируемых ядерными или митохондриальными генами. К настоящему времени идентифицирован ряд самостоятельных нозологических форм, связанных с мутациями митохондриальной ДНК.
Миоклонус-эпилепсия с разорванными красными мышечными волокнами (myoclonic epilepsy, ragged-red fibres - MERRF) впервые описана N. Fukuhara и соавт. в 1980 г. [2]. Заболевание обусловлено точечной митохондриальной мутацией гена транспортной РНК лизина, локализующейся у 80-90% больных в нуклеотидной позиции 8344. В отдельных семьях описана точечная мутация митохондриальной ДНК в нуклеотиде 8356 [3, 4]. Наследуется синдром МERRF по митохондриальному (материнскому, цитоплазматическому) типу с передачей потомству исключительно по материнской линии, при этом оба пола страдают с одинаковой частотой.
Патогенез болезни связывают с образованием аномальной транспортной РНК, которая способствует нарушению внутримитохондриальных процессов рибосомального синтеза и глубоким дефектам электронного транспорта, в частности, недостаточности I и IV комплексов дыхательной цепи [5, 6].
Клинические проявления болезни отличаются выраженным внутрисемейным полиморфизмом [7], который, по-видимому, связан с различным соотношением мутантной и нормальной митохондриальной ДНК в тканях организма [8].
Первые признаки заболевания могут появиться в возрасте от 3 до 65 лет. Клиническая симптоматика включает миоклонус, тонико-клонические судороги, атаксию, деменцию, нейросенсорную тугоухость, нерезко выраженный миопатический синдром, иногда атрофию дисков зрительных нервов. Редким проявлением патологии служит липоматоз [7, 9]. Болезнь характеризуется прогрессирующим течением.
Лабораторное обследование больных выявляет высокий уровень молочной кислоты в крови, увеличение содержания белка в ликворе, снижение активности I и IV комплексов дыхательной цепи в миоцитах скелетных мышц [10].
Важным морфологическим признаком патологии служит наличие в биоптатах скелетных мышц характерных <рваных> красных мышечных волокон ("ragged-red" fibres, RRF). Эти изменения указывают на скопление и пролиферацию аномальных митохондрий и типичны для целой группы митохондриальных энцефаломиопатий, обусловленных дефектами ДНК митохондрий. Помимо RRF, в миоцитах больных синдромом MERRF обнаруживаются гистохимические признаки недостаточности митохондриальных ферментов; электронно-микроскопический анализ выявляет нарушение количества и строения митохондрий, наличие цитоплазматических включений липидов, кальция и др. [10].
Лечение больных представляет исключительные трудности. В комплексе терапии предложено использовать препараты, повышающие эффективность электронно-транспортной цепи (коэнзим Q10, рибофлавин, никотинамид), антиоксиданты, противосудорожные средства. Однако положительное влияние такого лечения отмечено не у всех больных [11].
Мы наблюдали ребенка, страдающего синдромом MERRF с ранней манифестацией, выраженным прогрессирующим течением заболевания, тяжесть которого была обусловлена частыми генерализованными тонико-клоническими приступами и постоянными миоклоническими подергиваниями. У матери мальчика была диагностирована аналогичная патология. Своеобразие клинической картины болезни у матери заключалось в том, что, помимо типичных признаков синдрома MERRF, у нее были отмечены такие, ранее не зарегистрированные в литературе проявления, как обширные бородавчатый невус и гемангиома. В отечественной литературе описания семей с синдромом MERRF отсутствуют. В этой связи приводит наше наблюдение.
Больной Т., 12 лет, наблюдался в отделе врожденных и наследственных заболеваний Московского НИИ педиатрии и детской хирургии с 10-летнего возраста.
Жалобы при первом поступлении на мышечную утомляемость, тяжелые тонико-клонические судороги, сопровождающиеся потерей сознания, частые мышечные подергивания, нарушение школьной успеваемости, ослабление памяти.
Мать ребенка, 43 лет, предъявляла жалобы на повышенную утомляемость, слабость, мышечные боли, которые были особенно выражены в утренние часы. В школьном возрасте у женщины отмечались приступообразные мышечные подергивания (больше в нижних конечностях), что приводило к падению. При осмотре: физическое развитие ниже среднего (рост 156 см), окружность головы 54,5 см. Отмечалась асимметрия тела за счет гипотрофии мышц левых конечностей. Обращали на себя внимание бородавчатый невус, расположенный на внутренней поверхности левого бедра, и обширная гемангиома в области шеи, распространяющаяся на левое плечо. В неврологическом статусе: центральный парез VII и XII пар черепных нервов, выраженные симптомы орального автоматизма - ладонно-подбородочный, дистанционно-оральный, хоботковый. Миопатический симптомокомплекс умеренно выражен, усталость при длительной физической нагрузке, вставание из положения <на корточках> с применением вспомогательных <миопатических> приемов. Мышечная сила в проксимальных и дистальных группах умеренно снижена. Сухожильные рефлексы с двуглавой, трехглавой мышц плеча, коленные и ахилловы равномерно снижены (S = D). Умеренно выражены патологические пирамидные знаки - симптомы Бабинского, Оппенгейма. В позе Ромберга устойчива. Координаторные пробы - коленно-пяточную и пальценосовую - выполняла удовлетворительно. Чувствительные нарушения отсутствовали.
Отец ребенка, 36 лет, здоров. Среди других членов родословной отмечены случаи сердечно-сосудистой и онкологической патологии (рис. 1).
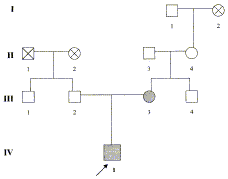 |
| Рис. 1. Фрагмент родословной семьи Т. I2 -инсульт, II1 - онкологическое заболевание, II2 - инсульт, III3 - синдром MERRF, IV1- синдром MERRF. |
Мальчик от 1-й беременности, протекавшей с токсикозом в первой половине и нефропатией, родов кесаревым сечением в 38 нед беременности. Масса ребенка при рождении 3100 г, длина 54 см, оценка состояния по шкале Апгар 8 баллов. На грудном вскармливании находился до 3 мес жизни. Раннее психомоторное развитие: голову держит с 3 мес жизни, сидит с 6 мес жизни, ходит с 1 года 2 мес.
В возрасте 2 лет впервые возникли судорожные пароксизмы по типу <кивков>. По этому поводу мальчик наблюдался невропатологом, получал лечение фенобарбиталом. Однако, несмотря на проводимую терапию, контроля над судорожными пароксизмами не наблюдалось, а отмечалось учащение и утяжеление приступов. С 7 лет появились генерализованные тонико-клонические пароксизмы, сопровождающиеся потерей сознания и остановкой дыхания. Трижды наблюдались эпизоды эпилептического статуса. Ребенок неоднократно обследовался в неврологических отделениях, получал массивное противосудорожное лечение, включающее финлепсин, реланиум, конвулекс, радедорм, синактен-депо. Несмотря на терапию состояние и самочувствие мальчика ухудшились, присоединились частые подергивания мышц конечностей и лица, усиливающиеся после физической нагрузки, частота миоклоний - до нескольких десятков в день. Нарушились память и внимание, резко снизилась школьная успеваемость. Ребенок был направлен на консультацию в Московский НИИ педиатрии и детской хирургии.
При поступлении состояние расценено как средней тяжести. В физическом развитии не отставал: индекс ДюРанта - Лайнера 92 (норма). Интеллектуальное развитие соответствовало умственной отсталости в степени легкой дебильности, IQ 72 ед. Кожные покровы чистые, бледные. В легких выслушивалось везикулярное дыхание. Тоны сердца ритмичные. Живот мягкий, безболезненный при пальпации. Аппетит, стул не нарушены. Зрение и слух не изменены.
Далее...
Написать комментарий
|
